



























Overview
Immunogenicity testing of therapeutic proteins is an essential step of the drug development process. Biological drug products often elicit an immune response in the patient. Clinical consequences of the presence of anti-drug antibodies (ADA) can vary from mild to serious adverse events. Therefore, the presence of ADAs is a major safety and efficacy concern and should be evaluated and correlated with any pharmacological or toxicological observations. The Alpha assay format provides a rapid and sensitive assay for the detection of anti-drug antibodies without the requirement of wash steps. AlphaLISA™ can be used to detect the presence of antibodies in a complex sample (serum, plasma, etc.), and is compatible with acid dissociation techniques used to dissociate immune complexes that can interfere with anti-drug antibody detection.
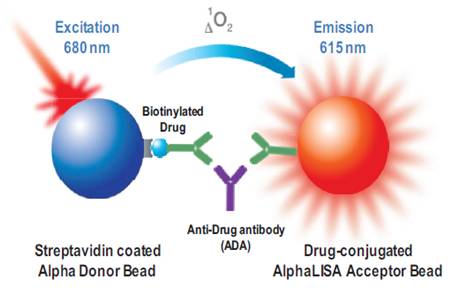
Detection of anti-drug antibodies (ADAs) can occur using various assay design formats (a bridging assay using labeled drug, or a direct capture assay using anti-IgG antibodies etc.). In the Alpha bridging assay, presence of the anti-drug antibody brings the Donor and Acceptor beads into proximity. Upon excitation of the Donor bead, ambient oxygen is converted to the excited singlet form of 1O2. If the Acceptor bead is within proximity of the Donor bead (~200 nm), the singlet oxygen can react with chemicals in the Acceptor bead to produce signal. This signal is proportional to the amount of anti-drug antibody present and can be quantified by running a standard curve.
Figure 1. Assay design for bridging immunoassay using biotinylated therapeutic antibodies.
What do I need to run this assay?
Required reagents available from Revvity:
- Alpha Donor beads appropriate for your assay
- Alpha Acceptor beads appropriate for your assay
- Microplates - We recommend our 96-well ½ AreaPlates or our 96-well and 384-well white OptiPlates™. Also see Microplate selection.
- TopSeal™-A adhesive plate seal for incubations
Instrumentation/equipment:
- A plate reader capable of reading Alpha assays (We recommend the Revvity EnVision™ or EnSight Plate Reader.)
Sample protocol-in-brief

Figure 2. Assay workflow
Assay development
View more detailed information on Alpha assay design and assay development.
Optimizations recommended for Alpha bridging assays:
- Titration of the biotinylated drug (from 0.01 nM up to 10 nM final in the well)
- Acid dissociation step: Acetic acid (600 mM) or Glycine-HCl (500 mM) pH 3.0 could be tested. The ratio of the neutralization buffer should be adjusted when different conditions of acid dissociation are tested to ensure that the neutralization is effective.
- Serum dilution factor,
- From ½ up to 1/20
- Buffer composition,
- Start with the recommended assay buffer (50 mM Tris-HCl pH 7.75, 0.5% BSA, 0.05% Bovine g-globulin, 0.1% Tween-20, 150 mM NaCl, and 10 mg/mL dextran). The Alpha Immunoassay buffer is generally not optimal for ADA assays.
- Time of incubation,
- 1h with the Acceptor beads for assays using the acid dissociation protocol.
- O/N with the Acceptor beads for the non-acid protocol.
In some instances, direct conjugation of the drug onto the Donor beads could give better results than drug biotinylation. For such situation, the following protocol could be used:
- In a 96-well OptiPlate dilute 5 µL serum samples with 20 µL Acetic Acid (600 mM).
- Incubate for 60 minutes in an incubator set at 23 °C.
- Add 75 µL of neutralization buffer (Tris-HCl 1 M pH 9.5 in assay buffer) containing 40 µg/mL drug-conjugated Donor beads and 40 µg/mL of AlphaLISA drug-Acceptor beads.
- Incubate for 60 minutes in an incubator set at 23°C.
- Read plate on an EnSpire or EnVision multi-label plate reader.
Assay qualification
Screening and confirmatory cut-point determination
Screening assays are used to determine the presence or absence of anti-drug antibodies in a sample. The screening cut-point is the signal of the screening assay at and above which a sample is considered to be reactive for the presence of antibodies. It can be determined statistically from a set of samples that are presumed to be anti-drug antibody-negative. Cut-point calculation typically yield a low level of false positive rate in order to decrease the risk of having false negative results. For this reason, confirmatory assays are needed to demonstrate that a positive result from the screening assay is a true positive (i.e. Sample containing a specific ADA). Various methods can be used to perform confirmatory assays, the most common being competitive drug inhibition assays (Smith et al.). In the competitive drug inhibition assay, positive samples are typically confirmed by spiking a known concentration of drug into the sample. True positive samples are confirmed if the percentage of inhibition observed in the spiked samples is higher than the confirmatory cut-point, which is determined statistically using a set of ADA-negative spiked samples.
In the data below, the screening cut-point (CP) was determined using 50 individuals lots of normal human serum, analyzed each in duplicate on a total of four occasions. The CP was calculated as the mean counts plus 1.645xSD. On each occasion, replicates of the pooled normal human serum (PNHS) were included and a correction factor was calculated, corresponding to the ratio between the CP and the mean counts of PNHS. Outliers (defined as lots with mean counts or % inhibition above the CP or CCP on more than 50% of the occasions) were removed for the 2nd iteration calculations. The 2nd iteration correction factor was used as a normalization factor for all assays in the qualification.
The confirmatory cut-point (CCP) was determined using the same 50 lots as for the CP, except that each lot was spiked with 25 µg/mL of the drug. The CCP was calculated as the mean of the percentage inhibition plus 2.33xSD. The LPC (low-level positive control) and HPC (high-level positive control) samples were included to confirm the CCP value. Outliers were also removed for the 2nd iteration calculations.
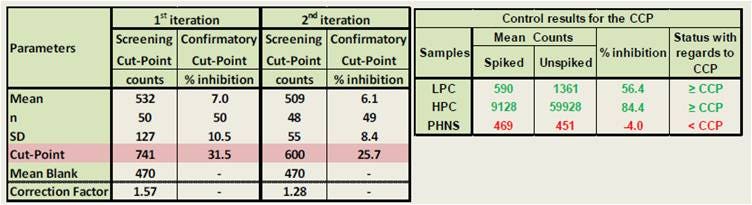
Figure 3. Determination of screening cut-point and confirmatory cut-point in an Alpha immunogenicity assay. For full protocol and details, refer to our poster.
Sensitivity and prozone
The assay should have an appropriate sensitivity (lower limit-of-detection) to detect clinically relevant levels of anti-therapeutic antibodies, from low affinity and high affinity antibodies. The assay should also have a good dynamic range, and not be affected by prozone effects. The prozone effect is a lack of detectable agglutination between antigen and antibody due to excess analyte. In Alpha assays, this can manifest itself in the form of a hook effect. One way to check for a prozone effect is to dilute samples further and compare interpolated anti-drug antibody concentrations.
In the data below, the pooled normal human serum (PNHS) was spiked with increasing concentrations of the positive control antibody to generate anti-drug antibody (ADA) standard curves. Highly intense signals and excellent sensitivity (4.9 ng/mL neat serum) were obtained. Three positive control (PC) concentrations were selected for the study (LPC, low level PC: 15 ng/mL, MPC, medium level PC: 250 ng/mL and HPC, high level PC: 1000 ng/mL). The sensitivity and prozone plate specific cut-point (PSCP) was calculated at 576 counts. The signal was still proportional to positive control antibody at a level 10-fold higher than the HPC, indicative of the high dynamic range of the assay.
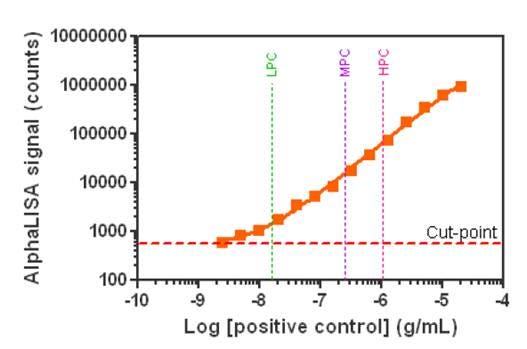
Figure 4. Determination of sensitivity in an Alpha immunogenicity assay. For full protocol and details, refer to our poster.
Drug tolerance
Samples containing circulating drug could exhibit assay interference as a result of the competition between the circulating drug in the sample and the drug used for association in the bridging assay. Compatibility of Alpha technology with acid dissociation techniques allows for improved drug tolerance.
In the data below, drug tolerance was tested. In order to mimic biological samples, the positive control antibody was diluted in PNHS and spiked with different amounts of drug. The samples were incubated 1 hour before analysis to allow for binding. Our results indicated that the assay could tolerate up to 20 µg/mL of free drug at the LPC level, whereas the HPC sample still produced a signal above the PSCP in the presence of 200 µg/mL of free drug. This result demonstrates that the AlphaLISA ADA detection assay exhibits an excellent drug tolerance.
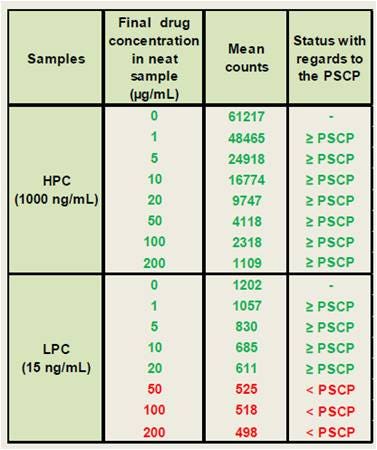
Figure 5. Drug tolerance in an Alpha immunogenicity assay.
Specificity and selectivity
Specificity is the ability of an assay to only score as a positive result if the sample contains an antibody that does indeed bind and/or neutralize the therapeutic protein/drug. "Positive" and "false positive" samples are distinguished during the confirmatory assay. On the other hand, selectivity is defined as the ability of an assay to measure the ADA independently of any possible matrix effect.
For the data shown below, the specificity was determined using ten lots of normal human serum. These individual lots were analyzed unspiked and compared to the PSCP level. All unspiked samples were below the PSCP level, indicating a good assay specificity. The selectivity was determined using the same lots of serum, except that each lot was spiked with the LPC or HPC concentrations. In addition, PNHS was spiked with the LPC and HPC levels to serve as reference for recovery determination. The mean counts were compared between individual serum lots and the reference samples. We observed 18 samples of 20 with count levels within a 25% difference of the corresponding PC prepared in PNHS, reflecting outstanding assay selectivity.
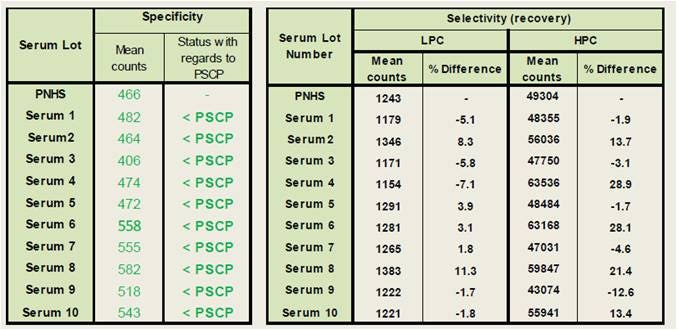
Figure 6. Determination of specificity and selectivity in an Alpha immunogenicity assay.
Reproducibility
Immunogenicity assays must be very reproducible, ensuring that a repeated assay on the same sample will produce the same result. For the data shown below, twelve independent experiments using triplicate data points were performed by two analysts using a balanced design to determine the reproducibility of the assay. The group mean, SD and coefficient of variation (%CV) of the mean counts obtained for each assay were calculated and used to calculate the intra-assay precision. The group mean, SD and %CV of all the intra-assay mean counts obtained over all occasions were calculated and used to determine the inter-assay precision. As shown in the table above, %CV values lower than 10% were obtained for all conditions, indicating a robust and reproducible assay.
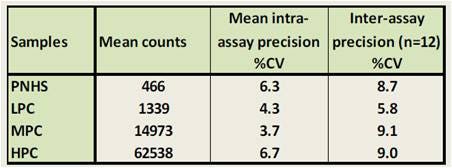
Figure 7. Determination of reproducibility in an Alpha immunogenicity assay. For full details, refer to our poster.
Acid dissociation
Acid-induced dissociation of immune complexes may be necessary to dissociate existing drug-antibody complexes within the sample to improve drug tolerance of the assay. Here is a sample protocol for acid-induced dissociation that has been tested in AlphaLISA immunogenicity assays:
- In a 96-well Optiplate dilute 5 µL serum samples with 45 µL Acetic Acid (600 mM).
- Incubate for 60 minutes in an incubator set at 23 °C.
- Add 50 µL of neutralization buffer (Tris-HCl 1 M pH 9.5 in assay buffer) containing 4 nM of biotinylated drug (in this case, the assay design includes biotinylated drug that will be associated with streptavidin-coated Donor beads) and 80 µg/mL of AlphaLISA drug-Acceptor beads.
- Incubate for 60 minutes in an incubator set at 23°C.
- Add 100 µL of 40 µg/mL streptavidin-Donor beads diluted in assay buffer
- Incubate for 60 minutes in an incubator set at 23°C.
- Read plate on an EnSpire or EnVision Multi-Label Plate Reader.
Tips
- Immunogenicity assays may require the use of an acid dissociation step prior to bead addition, to break existing drug-antibody interactions in your sample.
- Requirements for the drug tolerance will indicate if the acid dissociation step should be used. We recommend starting with 20 µg/mL of each bead in your reaction. Bead concentration can be titrated later in your assay development (in a cross-titration matrix, titrating each bead from 10 µg/mL to 40 µg/mL).
- The theoretical maximum capacity of a streptavidin-coated bead at 20 µg/mL of bead is 30 nM. Please note that this does not take into consideration the size of the biotinylated molecule that will associate with the bead. For example, you may find that a biotinylated antibody will saturate the bead at 2-3 nM, rather than at 30 nM. If you have saturated the bead, you may see a hook effect.
-
The theoretical maximum capacity of an antibody-coated bead is 3-10 nM. However, the hook effect typically does not happen until much higher concentration due to the nature of the antibody-antigen interaction.
For research use only. Not for use in diagnostic procedures.
The information provided above is solely for informational and research purposes only. Revvity assumes no liability or responsibility for any injuries, losses, or damages resulting from the use or misuse of the provided information, and Revvity assumes no liability for any outcomes resulting from the use or misuse of any recommendations. The information is provided on an "as is" basis without warranties of any kind. Users are responsible for determining the suitability of any recommendations for the user’s particular research. Any recommendations provided by Revvity should not be considered a substitute for a user’s own professional judgment.