



























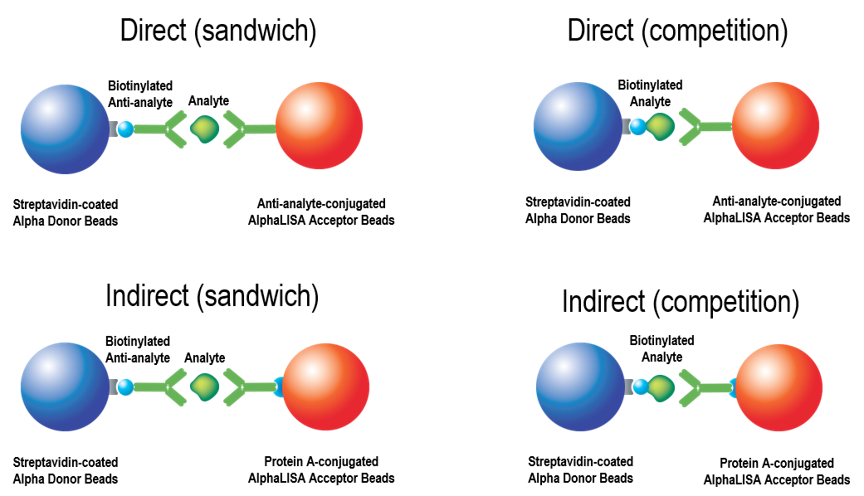
Overview
Many assays (analyte detection, antibody quantitation/characterization, post-translational modifications, kinase assays, protease assays, etc.) can be designed as immunoassays. Existing ELISAs can be converted to a no-wash AlphaLISA™ assay. Assays can be used for screening or for quantitation of an analyte (by running samples against a standard curve created with recombinant analyte). There are two major formats: a sandwiching antibody assay and a competition (displacement) assay. Each format can be performed using directly-conjugated beads (one antibody directly conjugated to an AlphaLISA Acceptor bead), or via an indirect method of antibody-bead association.
Fig. 1. Four basic configurations for Alpha immunoassays.
Sandwiching format
- The standard configuration is a sandwich assay employing two different antibodies that recognize non-overlapping epitopes on the target molecule.
- Increasing amounts of target in the sample result in higher luminescent signals. This is also referred to as a signal increase assay.
- The target molecule is not coupled to biotin.
Competition format
- The competition assay configuration is typically used when only one specific antibody is available to the target molecule.
- A purified source of the target molecule is labeled with biotin and added to the assay in a known concentration.
- Increasing amounts of unlabeled target in the sample compete with binding of the biotin-labeled target and result in a decrease in signal. This is also known as a signal decrease assay.
- Competition assays may have lower sensitivity than Sandwich assays, but they are not subject to hook effects at high analyte concentrations.
Direct configurations
- The Acceptor beads are directly coupled to the target-specific antibody.
- These assay configurations are often simpler to optimize and perform than the indirect configurations.
Indirect configurations
- The Acceptor beads are not directly coupled to the target-specific antibody. Instead, they are coupled to protein A or another antibody-binding moiety.
- The target-specific antibody is captured on Acceptor beads during an incubation step.
- This assay configuration is sometimes used when the target-specific antibody is available only in limited amounts (too small to allow a coupling reaction to beads).
- These assay configurations are somewhat more complicated to optimize and perform than the direct configurations.
- With competition assays, indirect assay configurations can often give equal or better sensitivity than direct assay configurations.
The most common format is the direct sandwiching assay, which uses a streptavidin-coated Alpha Donor bead and an antibody-conjugated AlphaLISA Acceptor bead. For sandwiching assays, it is necessary to have two antibodies that can simultaneously bind to your analyte. One antibody is biotinylated and will associate with the streptavidin-coated Donor bead. The other antibody is unlabeled and can either be directly conjugated to an AlphaLISA Acceptor bead, or can indirectly associate with a Protein A/Protein G/Anti-species AlphaLISA bead. With increasing amounts of analyte in your sample, the Donor and Acceptor beads will be brought together, resulting in an increase in signal when the Donor bead is excited.
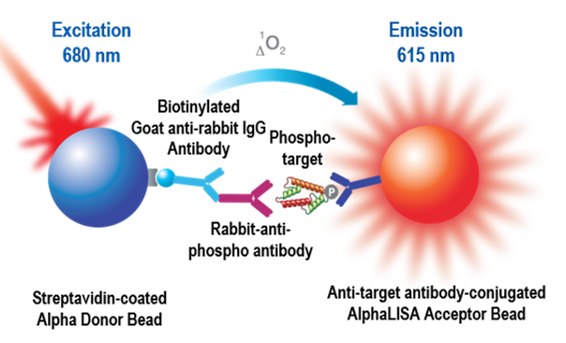
Fig. 2. Alpha sandwiching assay
If sandwiching antibodies are not available, or if your analyte is too small for a sandwiching assay, you should use the competition assay format. In the competition format, only one antibody is needed, and a biotinylated analyte is used as a probe for the assay. Your antibody is either directly conjugated to an AlphaLISA Acceptor bead, or indirectly associated with a Protein A/Protein G/Anti-species AlphaLISA bead. "Endogenous" unlabeled analyte will compete for binding to the antibody on the AlphaLISA Acceptor bead, displacing the biotinylated analyte and resulting in a decrease in signal.
What do I need to run this assay?
Required reagents available from Revvity:
- Alpha Donor bead (appropriate for your assay – usually a streptavidin Donor bead)
- AlphaLISA Acceptor bead (appropriate for your assay – usually an unconjugated AlphaLISA Acceptor bead. You would perform a bead conjugation with your antibody)
- Microplates - We recommend our 96-well 1/2 AreaPlates or our 384-well white OptiPlates™. Also see Microplate selection.
- TopSeal™-A adhesive plate seal
Required reagents available from other suppliers:
- Antibodies (For a sandwich assay format, usually one is biotinylated, while the other one is unlabeled.)
- Reagents for bead conjugation, if necessary
- Biotinylated probe (for a competition assay format)
- An appropriate buffer (we recommend our AlphaLISA ImmunoAssay Buffer.)
Instrumentation/Equipment:
- A plate reader that is capable of reading Alpha assays (e.g. EnVision™ or EnSight™ Reader)
Assay development
Detailed information for each optimization step.
- Determining the best antibody pair
- Assay buffer optimization
- Biotinylated antibody titration
- Order of addition
- Incubation time
- Bead ratio
- Determining a suitable matrix (diluent) for your standard curve
- Tips for competition assay formats
FAQs
Q. What is the main difference between AlphaLISA and AlphaScreen assays?
A. Both assay formats use the same type of Donor bead. The main difference between the two assays is in the Acceptor beads. With AlphaScreen Acceptor beads, the final light emission is from rubrene; with AlphaLISA Acceptor beads, the final light emission is from Europium. The emission wavelength is different between AlphaScreen beads (520-620 nm) and AlphaLISA beads (615 nm). Because of the narrow emission spectrum of AlphaLISA beads, there is much less interference in serum and plasma samples, and better sensitivity is obtained. Additionally, if you are screening small molecule libraries, you might want to consider using AlphaLISA beads to reduce interference from small molecules that absorb between 520 and 600 nm.
Q. Can I use Protein A-coated beads in my assay design?
A. If you are using two antibodies, you will need to make sure that only one of your antibodies can bind to Protein A. Pierce has a nice chart that can be helpful - you would want one antibody that binds strongly with Protein A, and one antibody that either doesn't bind (or binds weakly) to Protein A. If both your antibodies could bind well to the same bead, you would have high assay background. We also carry anti-species beads to capture antibodies (if your antibodies are from different species). Note that if you are using antibodies as test compounds in your assay, you will probably want to stay away from Protein A beads.
Q. Can I have both the Donor and Acceptor beads directly-conjugated to my antibodies?
A. You could, but in general, you will get the best assay sensitivity when using one directly-conjugated bead and one biotinylated antibody with a streptavidin-coated bead. This allows you to play with order of addition as well.
Q. Can I use the same antibody on both beads?
A. Yes. This works best when using a polyclonal antibody, though. It can work for a monoclonal antibody as well, but here you are going to detect only homodimers. The latter could be a nice strategy for monitoring aggregation or dimerization. However, if you are monitoring aggregation or dimerization, the Alpha assay won’t be “quantitative”, meaning that a single pair or a big aggregate of hundreds of protein will give the “same” signal (signal from one bead pair). As for how to associate the same antibody to the two beads, there are various possibilities. The most classic one is to use a biotinylated version of the antibody with streptavidin-coated Donor beads and direct conjugation of the unlabeled version of the antibody on Acceptor beads. Double indirect association of the antibody (for example, using Protein A Donor beads and anti-rabbit IgG Acceptor beads to capture the same rabbit IgG antibody on both sides) would be trickier as it is possible a single rabbit IgG antibody might bridge a Donor and Acceptor bead in the absence of any other reagents, and is currently being evaluated.
Q. If I need to use a competition format, what concentration of probe/tracer should I use?
A. We would recommend testing between 0.5 nM and 10 nM probe (with 20 µg/mL of each bead). Lowering the concentration of your probe will make the assay more sensitive.
Q. Can I use monoclonal antibodies on both sides of the reaction?
A. As long as the antibodies recognize non-sterically competing epitopes, you can use two monoclonal antibodies.
Q. Is there a way to predict what antibody pairs might work?
A. We recommend that you determine this experimentally, as antibody selection can have a dramatic impact. Sometimes you can find sandwiching pairs from various vendors – pairs of antibodies that have already been shown to work in sandwiching assays. For example, some providers will give you pair info (e.g. BD Biosciences, MBL, Biolegend, and BioPorto sometimes give this information on their antibody TD sheets). If someone has published a paper where they have developed a home-made ELISA, you can sometimes find antibody pair info in the details of the Material and Methods section.
Q. Since I have a western blot that works, will these antibodies also work for my Alpha assay?
A. Antibodies successfully used in western blot analysis are more likely to work in AlphaLISA. Having said that, this is no guarantee of success. Contact your Revvity Drug Discovery Specialist for advice on the antibody selection process.
Q. What assay buffer should I use for my assay?
A. The assay buffer refers to the buffer used to dilute your Donor beads, Acceptor beads, and antibodies. We offer multiple buffers for AlphaLISA immunoassays. Most assays will work best using 1X AlphaLISA Immunoassay Buffer (Cat. No. AL000) for dilution of your beads and antibodies. For assays exhibiting high background, AlphaLISA HiBlock Buffer (Cat. No. AL004) or AlphaLISA NaCl Buffer (Cat. No. AL007) could be tested.
| Assay buffer | Catalog number | Key buffer components | Recommended for: |
|---|---|---|---|
| 1X AlphaLISA Immunoassay Buffer | AL000C/F | HEPES buffer pH 7.4, casein, Dextran-500, Triton X-100 | Most AlphaLISA immunoassays |
| 1X AlphaLISA HiBlock Buffer | AL004C/F | HEPES buffer pH 7.4, casein, Dextran-500, Triton X-100, BSA, and gelatin | Optimized assays that exhibit high background due to sample matrix |
| 1X AlphaLISA NaCl Buffer | AL007C/F | High concentration of NaCl with HEPES buffer pH 7.4, casein, Dextran-500, Triton X-100, and gelatin | Optimized assays that exhibit high background due to sample matrix, but where it is not allowable for BSA to be present in the buffer (alternative to AL004) |
Q. What should I use to dilute my analyte, for my standard curve?
A. During the first steps of assay development, AlphaLISA Immunoassay Buffer (Cat. No. AL000) can be used to dilute your analyte. However, once you have a working assay, you will need to use a diluent for your standard curve that will closely match your sample matrix. For example, if you are working with serum samples, you might use FBS to dilute your analyte for your standard curve. Linearity and spike-and-recovery experiments should be performed to validate your standard curve diluent. For more information, refer to our section on& Working with serum and other complex samples.
Q. What is the binding capacity of the beads?
A. 1 µg of beads is 2x10^[8] beads. Each bead can bind 200-300 160 kD antibody molecules. At 20 µg/ml of beads, that equates to 2-3 nM antibody.
Q. How much sample volume can be used for an AlphaLISA assay?
A. The standard sample volume is 5 µl in a 50 µl reaction, but the reactions performed in buffer can be adapted to allow the addition of up to 20 µl of sample to increase sensitivity. For reactions performed in sample matrices other buffer, the maximum sample volume should be determined for each analyte.
Q. How much total volume of serum can be used in the assay?
A. Due to the fact that the hemoglobin in the serum will capture some of the oxygen released by the Donor beads, the percentage of serum should not exceed 10% of the total volume (i.e. 5 µl serum sample in a 50 µl final assay volume). Make sure to choose AlphaLISA Acceptor beads when working with serum.
Q. I am working with serum samples. How do I prepare analyte-depleted serum to use for my standard curve?
A. We have a protocol for preparing analyte-depleted serum. If you are preparing standards for quantitation and will be working with serum samples, you will need to use analyte-depleted serum to dilute your recombinant standard.
Q. I am measuring a biomarker in a cellular model. Can I perform the AlphaLISA assay directly in a cell culture plate and avoid the transfer step?
A. Yes. AlphaLISA assays generally can be performed in the presence of cell culture media and cells. Minor optimization might be needed depending on your cellular model. Refer to our Working with cell extracts and supernatants in Alpha assays page for more information.
For research use only. Not for use in diagnostic procedures.
The information provided above is solely for informational and research purposes only. Revvity assumes no liability or responsibility for any injuries, losses, or damages resulting from the use or misuse of the provided information, and Revvity assumes no liability for any outcomes resulting from the use or misuse of any recommendations. The information is provided on an "as is" basis without warranties of any kind. Users are responsible for determining the suitability of any recommendations for the user’s particular research. Any recommendations provided by Revvity should not be considered a substitute for a user’s own professional judgment.