



























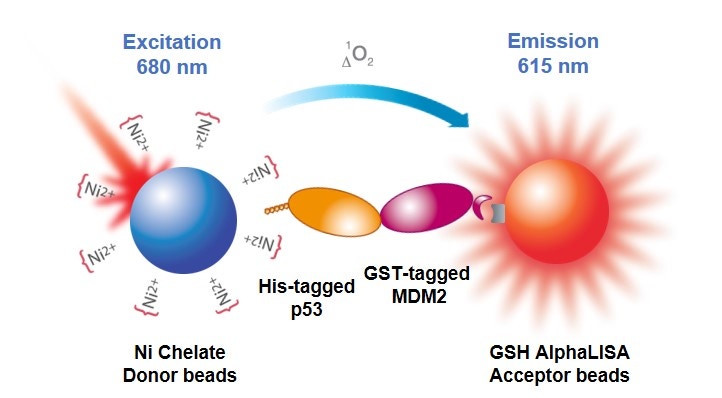
Overview
The use of tagged proteins, peptides, and other reagents (6xHis, GST, c-myc, HA, digoxigenin, etc.) can be particularly useful when studying biomolecular interactions or enzymatic activity with recombinant or synthetic substrates (kinase assays, protease assays, helicase assays, etc.). The use of a tagged reagent with an appropriate anti-tag bead or affinity bead can eliminate the need for an antibody in your assay design.
Fig 1. Alpha protein-protein interaction assay using tagged reagents.
What Do I Need To Run This Assay?
Required reagents available from Revvity:
- Alpha Donor beads, appropriate for your assay
- AlphaScreen™ or AlphaLISA™ Acceptor beads, appropriate for your assay
- Microplates - We recommend our 96-well 1/2 AreaPlates or our 384-well white OptiPlates™
- TopSeal-A™ adhesive plate seal for incubations
Instrumentation/equipment:
- A plate reader (capable of reading Alpha assays)
- Optional: plate rocker
Assay Design
Tags
A. Non-peptidic
Digoxigenin (DIG), biotin, and fluorescein/FITC are all tags that can be used with our beads. These tags are usually added exogenously to proteins and peptides, via chemical coupling reactions. Revvity provides protein and peptide biotinylation, as well as FITC labeling custom services. These tags can also be incorporated into nucleic acids by using a labeled nucleotide analog during oligo synthesis. For a list of Revvity nucleotide analogs, view our hapten nucleotides. Other companies provide oligo custom synthesis with DIG, biotin, and fluorescein/FITC modifications. Depending on your labeling method, it can be hard to control the number of tags per molecule. You should consider if your oligos or peptides need to be end-labeled for your assay. If you find that you need to biotinylate a protein, there are essentially two approaches to add biotin to a full-length protein. The first method is by chemical reaction (for example, biotinylation using SoluLink NHS-biotin). The second method is by engineering a recombinant form of the protein encoding a biotinylation sequence that may be labeled in vivo or in vitro (for example, using the Biotin AviTag™ system from Avidity, LLC). The latter choice offers a more-specific labeling of the binding partner.
B. Peptidic
Peptidic tags, such as 6xHis, GST, c-Myc, FLAG, and HA, can all be used with our beads. These tags are usually encoded into the recombinant expression plasmid or incorporated during peptide synthesis. These tags vary in length, from several amino acids to 100+ residue proteins. You can position the tag at the C-terminus, N-terminus, or within the amino acid sequence of your protein as desired to prevent steric hindrance of your binding interactions.
Bead Capacity
There is a limited capacity on the bead. If you exceed the capacity of the system, you might see a hook effect. As an example, the amount of glutathione on the surface of a bead is more than that of anti-GST antibody. But you would still have to consider how much GST-tagged protein could interact with either bead. Because the interaction of the GST tag with glutathione is weaker, you can add more GST-tagged biomolecule before saturating the glutathione bead. Since the interaction of the GST tag with an anti-GST antibody is stronger, you would reach the hook point more easily than with a glutathione bead.
- The theoretical maximum capacity of a streptavidin-coated bead at 20 µg/mL of bead is 30 nM. Please note that this does not take into consideration the size of the biotinylated molecule that will associate with the bead. For example, you may find that a biotinylated antibody will saturate the bead at 2-3 nM, rather than at 30 nM. If you have saturated the bead, you may see a hook effect.
- The theoretical maximum capacity of an antibody-coated bead is 3-10 nM. Please note this is a theoretical number --- you may be able to add more or less than 3-10 nM "antigen" before saturating the bead, depending on how strong the antibody-antigen interaction is, and how large your antigen is.
- The theoretical maximum capacity of a Ni-NTA or glutathione bead for a tagged protein is ~ 30 nM. However, because the His-Ni-NTA and GST-glutathione interactions are weak compared to other interactions (and more tagged protein will be dissociated from the bead at equilibrium), you will likely be able to use 300-1000 nM tagged protein before reaching the hook point.
- Bead capacity chart
Equilibrium
For a biomolecular interaction assay, you will need a certain amount of AB interaction for signal.
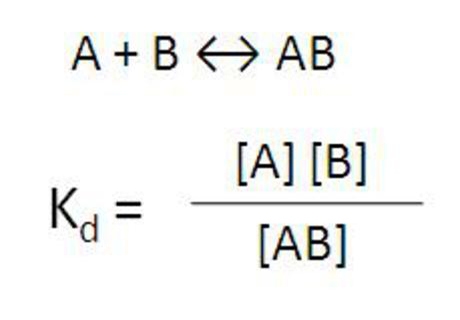
Fig. 2. Equilibrium of binding of molecule A to molecule B.
If the interaction between A and B is strong (as in an antibody-analyte interaction), there will be lots of AB complex. If the interaction between A and B is weaker, as in most protein-protein interactions or a glutathione-GST or Ni-NTA-His tag interaction, you may need to increase concentration of one protein or the other to drive the equilibrium towards more AB complex or increase the amount of both proteins to generate more AB complex.
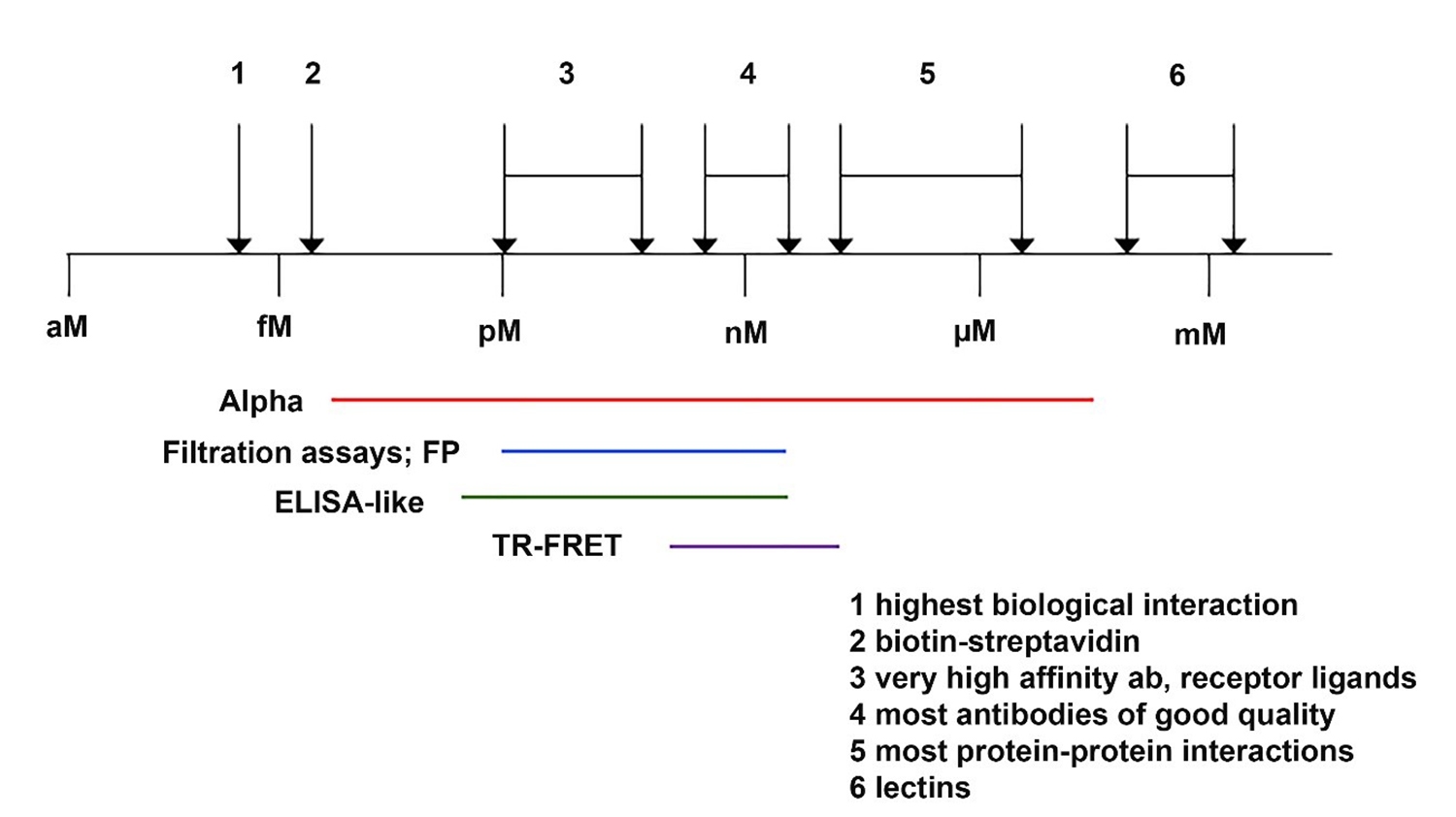
Fig 3. Binding affinities for various interactions.
Affinity
A. For streptavidin-coated bead + biotinylated component
- Pros: The binding between streptavidin and biotinylated component is very strong, so most of the biotinylated component will be associated with the bead, rather than being free in solution.
- Cons: You can exceed the bead binding capacity more quickly because the binding is so strong.
B. For antibody/protein A bead + protein:
- Pros: The binding between antibody and protein is fairly strong, so most of the antibody will associate with your protein.
- Cons: You can exceed the binding capacity more quickly because the binding is fairly strong.
C. For anti-GST bead + GST-tagged protein:
- Pros: The binding between antibody and tagged protein is fairly strong, so most of the antibody will associate with your tagged protein.
- Cons: You can exceed the binding capacity more quickly because the binding is fairly strong.
D. For Glutathione bead + GST-tagged protein:
- Pros: This affinity is weaker, so you won't exceed the binding capacity as quickly; you can add more GST-tagged protein if you need to push the binding equilibrium to AB complex.
- Cons: The affinity is weaker, so there will be a larger proportion of GST-tagged molecules that aren't associated to bead.
E. For competition assays, using a biotinylated analyte as a tracer is usually best.
- The biotin-streptavidin interaction is strong, so you can use less biotinylated tracer and usually still get good signal (most of the biotinylated tracer associates with antibody-associated bead and streptavidin bead rather than being unassociated).
- Using a lower amount of tracer will make your assay more sensitive (less probe = more sensitive assay).
Competition Assays
- Competition assay formats (displacement assays) are often used when sandwiching antibodies are unavailable. Here are some general tips for competition assays:
- Try an indirect assay format in which the antibody is not directly coupled to the Acceptor bead, but rather is captured by a Protein A Acceptor bead. This approach usually gives higher total counts and equal or better sensitivity.
- If you are using a biotinylated tracer/probe, the linker length of the biotinylated analyte probe can be important in developing a successful assay. To overcome steric hindrance to antibody binding, some assays (but not all) require that a 6-carbon chain be used to link biotin to the analyte.
- If you are using a competition (displacement) format, we would recommend testing between 0.5 nM and 10 nM probe (with 20 µg/mL of each bead). Lowering the concentration of your probe will make the assay more sensitive.
- The order of addition should allow binding of the analyte sample to the antibody before other assay components are added. Try a 3-step protocol:
- Add analyte sample to antibody (or antibody-coupled Acceptor beads). Incubate for 30 min.
- Add tagged analyte probe. Incubate for 30 min.
- Add affinity-Donor beads (and Protein A-coupled Acceptor beads, if doing an indirect assay). Incubate for 30 min.
Buffer selection
Visit Buffer selection for Alpha assays for help in choosing a suitable buffer for your assay. This page describes the various Alpha immunoassay buffers supplied by Revvity and will help you avoid interfering substances in buffers of your own design.
Initial Experiments
1. Determining optimal reagent concentrations
Choose a suitable buffer system and titrate each binding partner to ascertain the optimal concentration. It may also be necessary to vary the order of addition of the components to permit the most efficient interactions. For initial experiments, we recommended adhering to a final bead concentration of 20 µg/mL for both Donor and Acceptor beads. Subsequence titration of the beads (cross-titrating the Donor and Acceptor bead concentrations from 10 µg/mL to 40 µg/mL) may be assessed once it is known that a sufficiently high signal-to-background (S:B) can be achieved. Typically, most Alpha assays will use a final biotinylated binding partner concentration in the nanomolar range (0.5-30 nM with 20 µg/mL of beads). Concentration ranges for each binding partner that interact directly with the capture molecule on the AlphaScreen beads are usually between the low nanomolar range up to high nanomolar range (0.1-300 nM), depending on the affinity of the binding partners and the efficiency of labeling and/or stoichiometry of the capture tag/epitope (again working at 20 µg/mL of beads). See the example below:
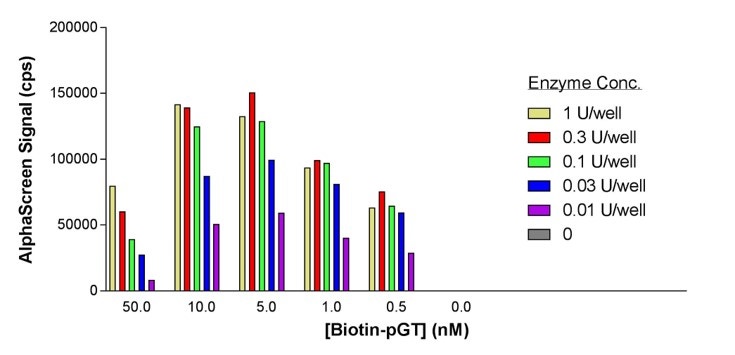
Fig 4. Optimization of c-Src tyrosine kinase enzyme and biotin-poly-GT substrate concentrations.
- The theoretical maximum capacity of a streptavidin-coated bead at 20 µg/mL of bead is 30 nM. Please note that this does not take into consideration the size of the biotinylated molecule that will associate with the bead. For example, you may find that a biotinylated antibody will saturate the bead at 2--3 nM, rather than at 30 nM. If you have saturated the bead, you may see a hook effect.
- The theoretical maximum capacity of an antibody-coated bead is 3-10 nM. Please note this is a theoretical number; you may be able to add more or less than 3-10 nM "antigen" to your assay before hooking, depending on how strong the antibody-antigen interaction is and how large your antigen is.
- The theoretical maximum capacity of a Ni-NTA or glutathione bead for a tagged protein is 3-30 nM, depending on the size of your protein. However, because the His-Ni-NTA interaction and the GST-glutathione interaction are weak (relative to biotin:streptavidin interaction, for example), you may be able to use more than 30 nM tagged protein (possibly up to 100 nM or more) before saturating the bead.
- If you are using a competition (displacement) format, we would recommend testing between 0.5 nM and 10 nM probe (with 20 µg/mL of each bead). Lowering the concentration of your probe will make the assay more sensitive.
2. Generating data
Assay development times for Alpha assays are typically very short due to the rapid speed with which one can generate data. It is not uncommon to have the basic format and preliminary conditions for a new assay determined within a day or two using Alpha. In part this is possible due to the very high signal-to-background ratios (S:B) that are typical for many Alpha assays, and hence the wide range of suitable conditions with which to generate a signal. In addition, most assays may be prepared and ready to read within 3 hours.
Following initial reagent component optimization and buffer selection, EC50 values and IC50 values as relevant can be determined very easily.
Data output from the EnVision™ with Alpha module, VICTOR Nivo™, or EnSight™ can be generated in various formats which are easily transferred to data analysis programs such as Excel or GraphPad Prism.
3. Interpreting data
Analysis and interpretation of data should be done with the analysis program of choice. Alpha assays are typified by having very low variability between replicate sample wells. It is often enough to run assays in duplicate. Alpha can be used to screen samples in singlicate. Alpha typically has extremely high Z' factors (0.8--0.9).
Optimizing Your Assay
1. Assay format
Assay format is an important consideration and can greatly impact the performance of the assay, the throughput, and the cost per well. The number of additions, the time taken to aliquot the reagents, the capacity and speed of the automated liquid handling robot (if the assay is ultimately intended for HTS) should all be taken into account when choosing the optimal assay format. Assay development procedures are frequently run using 96- or 384-well microplate formats, often with the use of a multi-channel pipette. The observed Alpha signal will be influenced by plate type and density.
Some plates, such as the ProxiPlate™ or our 1536-well plates, are designed to place the sample closer to the detector and therefore will give an increased signal. Also, the measured signal is dependent upon reflected light, therefore the reflective properties of the plate influence the signal. Higher density plates possess narrower wells that more-efficiently reflect the emitted light back to the detector. The width of the laser beam in our instruments is 1 mm, which is the same for all plate densities. Therefore, there is an effective reaction volume (the portion of the well hit by the laser light directly), as well as a total well volume. Higher density microplates allow for a higher proportion of the total well volume to be illuminated, hence greater signal generation.
Alpha is intended to be used as a homogeneous assay and does not require wash steps. However, this does not preclude the user from running a non-homogeneous format assay. For example, you may run an enzymatic reaction at a high substrate concentration due to Km restraints, then dilute an aliquot of the mixture into an Alpha reaction in the well. This may be necessary if the sensitivity of the assay far exceeds the range in which the biological reaction must be run.
2. Order of addition
Order of addition can influence the signal to a large extent. The optimal order in which assay components interact should always be determined empirically. You should keep in mind that some binding partners may abrogate the association of others if allowed to interact in the wrong order. This is seen in the example of a sandwich immunoassay, where steric hindrance by one antibody prevents the other antibody from binding. In general, one nearly always gives the advantage of the antibody-antigen interaction over the biotin-streptavidin association. One notable exception to this rule is the AlphaScreen cAMP assay, which uses a biotinylated cAMP as a probe in a competition assay. Biotinylated cAMP is a relatively small molecule, and if exposed to the antibody before being bound to the streptavidin Donor bead, a noticeable reduction in signal is observed due to a proportion of the biotin molecules being "smothered" by the antibody. In addition, biotinylated cAMP is pre-bound to the streptavidin Donor bead, as free streptavidin can interact with components in the cell lysate. Furthermore, free biotin present in the cell culture medium or in the bacterial culture can also lead to reduction of the Alpha signal, when using streptavidin beads.
3. Reagent choice
Reagent choice is critical. Where possible, using purified binding partners is always preferable. Affinity-purified polyclonal (pre-adsorbed if necessary) and/or monoclonal antibodies should be considered first. Importantly, the method of assay component capture to the bead has to be selected with the molecules of interest in mind. One should choose a suitable method of assay component capture, compatible with the nature of the assay and the required dynamic range of the assay.
4. Buffer choice
Buffer choice can be very important. Choose the pH, buffering capacity, and salt concentration that will facilitate the desired interactions between the components of your assay. If metal co-factors are needed for correct conformational integrity or enzymatic activity, it is best to titrate these components appropriately. In the case of excessive non-specific binding, a variety of different detergents, such as Tween-20 (0.01-0.1%), Triton X-100 (0.01-0.1%), or CHAPS (0.1% or less) may be used. For most Alpha applications, a BSA concentration of 0.1% (w/v) is sufficient to minimize non-specific interactions. Some assays may require slightly higher concentrations of BSA or even the use of an alternate blocking reagent such as low molecular weight dextran or gelatin. Try to avoid azide as a preservative, as this is a potent scavenger of singlet oxygen and will inhibit the Alpha signal. Proclin® 300 (Sigma-Aldrich) is recommended as a preservative/anti-microbial agent.
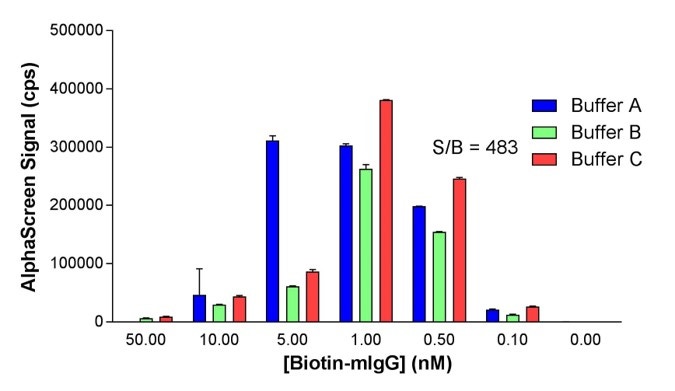
Fig. 5. For the optimization of an IgG detection assay shown above, it is clear that the different buffer components can influence the optimal binding partner concentration. Buffer C yields the highest signal and also permits the use of a lower concentration of biotin-mouse-IgG compared to buffer A. Buffer conditions will affect S:B but can also significantly influence the sensitivity and dynamic range of an assay.
5. Salts, detergents, and ions
The use of different salts and detergents in assay buffer systems can significantly affect the specific biological interactions of interest and the performance of the assay. If a low maximum signal window or high background is apparent, consider trying a different buffer, type of salt, salt concentration, reducing agent, or detergent.
We strongly recommend avoiding the use of transition metal ions: Al2+, Fe2+, Fe3+, Cu2+, Ni2+, and Zn2+. These metals have been shown to be potent singlet oxygen quenchers in the mM and sub mM ranges (100 µM for Fe2+).
Tips and FAQs
- If you will be using antibodies as inhibitors or antagonists in your reaction (antibodies to break an interaction, antibodies to block a receptor), you probably want to stay away from Protein A beads.
- For protein-protein interaction assays, you might consider using weaker affinity beads (for example, Ni-NTA and glutathione beads, rather than anti-GST and anti-His antibody beads) to bind your tagged biomolecules. There is less risk of hooking on the weaker affinity beads.
- We recommend starting with 20 µg/mL of each bead in your reaction. Bead concentration can be titrated later in your assay development (in a cross-titration matrix, titrating each bead from 10 µg/mL to 40 µg/mL).
- The theoretical maximum capacity of a streptavidin-coated bead at 20 µg/mL of bead is 30 nM. Please note that this does not take into consideration the size of the biotinylated molecule that will associate with the bead. For example, you may find that a biotinylated antibody will saturate the bead at 2-3 nM, rather than at 30 nM. If you have saturated the bead, you may see a hook effect.
- The theoretical maximum capacity of an antibody-coated bead is 3-10 nM. Please note this is a theoretical number; you may be able to add more or less than 3-10 nM "antigen" to your assay before seeing a hook effect, depending on how strong the antibody-antigen interaction is.
- The theoretical maximum capacity of a Ni-NTA or glutathione bead for a tagged protein is 3-30 nM, depending on the size of your protein. However, because the His-Ni-NTA interaction and the GST-glutathione interaction are weak (relative to biotin:streptavidin interaction, for example), you may be able to use more than 30 nM tagged protein (possibly up to 100 nM or more) before saturating the bead.
- If you are using a competition (displacement) format, we would recommend testing between 0.5 nM and 10 nM probe (with 20 µg/mL of each bead). Lowering the concentration of your probe will make the assay more sensitive.
- Alpha immunoassay design.
Q. Can I use the same bead coating for both the Donor beads and the Acceptor beads in my assay design?
A. Yes, it is possible to use the same tag on both sides. There are two risks associated with this approach. First, the tag must be unique on each protein (i.e., one tag per protein). For example, a poly-biotinylated protein would not be ideal to use in this format, as a single poly-biotinylated protein could potentially bridge a streptavidin-coated Donor bead and a streptavidin-coated Acceptor bead, leading to a false positive signal. Second, there is also the risk of detecting not only protein 1-protein 2 complexes, but also protein 1-protein 1 or protein 2-protein 2 complexes (homodimers). Controls are therefore very important - each protein should be tested in the assay individually, to ensure no significant signal comes from either protein forming homodimers.
For research use only. Not for use in diagnostic procedures.
The information provided above is solely for informational and research purposes only. Revvity assumes no liability or responsibility for any injuries, losses, or damages resulting from the use or misuse of the provided information, and Revvity assumes no liability for any outcomes resulting from the use or misuse of any recommendations. The information is provided on an "as is" basis without warranties of any kind. Users are responsible for determining the suitability of any recommendations for the user’s particular research. Any recommendations provided by Revvity should not be considered a substitute for a user’s own professional judgment.