




























The concept of targeted protein degradation challenges conventional rules for pharmaceutical drug discovery. Rather than directly modify protein function by occupying an active site, PROteolysis TArgeting Chimeras (PROTACs) exploit the cell’s own degradation machinery to irreversibly destroy target proteins. Not only is this novel strategy enabling development of more potent and selective treatments for cancer and other intractable diseases, it is also unlocking parts of the proteome that have traditionally been considered “undruggable”.
A new class of weapon for pharmaceutical drug discovery
Today’s precision molecular therapies are dramatically improving outcomes for patients suffering from complex diseases such as cancer. However, by design, the vast majority of pharmaceutical drugs are like single-use bullets, each drug molecule capable of taking out just one target protein. By analogy, PROteolysis TArgeting Chimeras (PROTACs) are more like designer bullets that can be used over and over again. Each PROTAC molecule can seek out and destroy many protein molecules in its lifetime. This unique mechanism of action makes PROTACs a fundamentally different class of weapon, with novel physicochemical properties not offered by other drug modalities.
What are PROTACs?
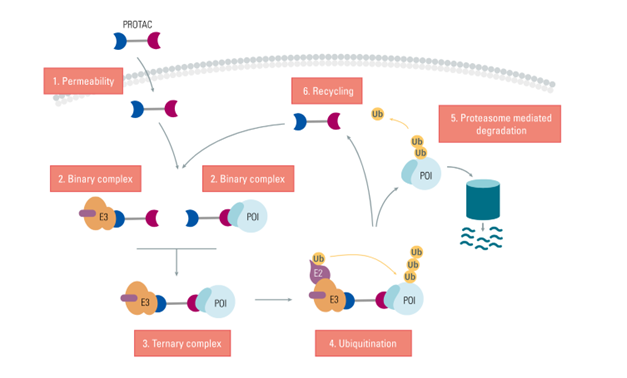
PROteolysis TArgeting Chimeras (PROTACs) are small two-headed (heterobifunctional) molecules that hijack the cell’s protein degradation machinery to irreversibly destroy the target. Each PROTAC consists of a linker with a ligand on one end to confer target specificity, and a different ligand on the other end to engage the cell’s ubiquitin system. The PROTAC thus acts as a bridge that brings the target of interest into close proximity with an E3 ubiquitin ligase (Fig 1). This enables the E3 ligase complex to catalyze ubiquitination of the target. The resulting polyubiquitin chain flags the target protein for degradation by the proteasome.
Figure 1 : PROTAC mechanism. Each heterobifunctional PROTAC molecule targets a protein of interest (POI), serving as a bridge to bring it into close proximity with an E3 ligase. The resulting ternary complex catalyzes polyubiquitination of the target protein, tagging it for degradation by the proteasome.
PROTACs and cancer therapy
While PROTAC technology has many potential application areas, by far the biggest focus to date has been the development of novel drugs for cancer therapy.
Protein kinases have become one of the most important target classes for the development of more effective anticancer drugs. Kinases play key roles in signal transduction and regulation of diverse pathways essential for vital cell functions such as gene expression, cell cycle progression, metabolism, cell motility and apoptosis. Since many of these same pathways can be subverted to enable uncontrolled proliferation, metastasis, and escape from immune surveillance, it is not surprising that dysregulation of kinase activity has been implicated in many types of cancer.
Over the past decade there has been an explosion in the number of small molecule inhibitors targeting protein kinases that regulate key cancer-related pathways and signal transduction cascades. Since FDA approval of the BCR-ABL kinase inhibitor imatinib (trademarked Gleevec) in 2001, the FDA has approved 62 small molecule protein kinase inhibitors 1. The Protein Kinase Inhibitor Database currently lists over 250 small molecule PKIs in clinical trials 2.
In search of better small molecule drugs
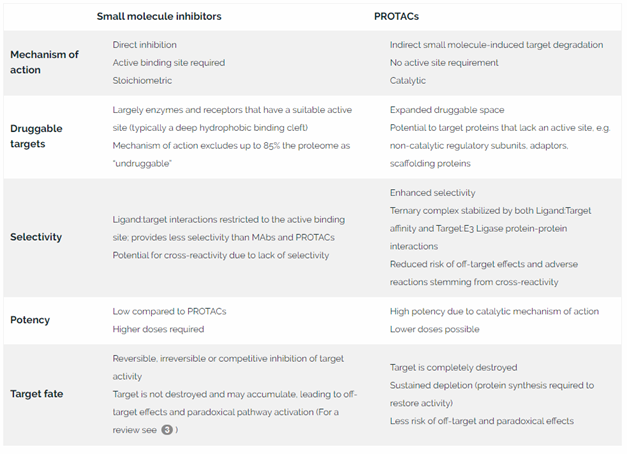
Table 1. Comparison of PROTACs with traditional small molecule inhibitors
The PROTAC advantage
Due to its unique mechanism of action, PROTAC technology offers significant advantages as an alternative to small molecule inhibitors:
No active site requirement
Unlike conventional inhibitor drugs, PROTACs do not require a deep hydrophobic binding pocket or active site. This permits development of novel ligands that can bind to other areas of the target protein. It also expands the druggable proteome beyond enzymes and receptors, to include new target classes that do not have classical ligand binding sites, such as adaptor molecules, non-catalytic regulatory subunits and scaffolding proteins.
High potency
Because PROTACs flag target molecules for degradation but are not themselves consumed in the process, each PROTAC molecule can act over and over again to destroy many disease-causing proteins during its lifetime. This catalytic mechanism of action means that PROTACs have the potential to be much more potent than inhibitor drugs designed based on a site occupancy model.
Exceptional selectivity
PROTAC activity depends not just on target:PROTAC affinity, but also on protein-protein interactions between the target and the E3 ligase, which confer stability to the ternary complex. This provides an extra tier of selectivity, which may reduce the risk of off-target side effects and adverse reactions. In several instances, PROTACs have demonstrated higher affinities than the inhibitor ligand on its own 4.
Sustained target depletion
PROTACs can mediate rapid target depletion, eliciting measurable responses within a few hours 5. Because PROTACs mediate the irreversible destruction of target proteins, protein synthesis is needed to restore target activity, and the effect is sustained for as long as 48 hours after exposure 5. An additional benefit of complete destruction of the target protein is that compensatory side effects, such as paradoxical activation of the targeted pathway, are less likely.
Potential to overcome drug resistance
Cancers often develop resistance to small molecule inhibitors by developing mutations at the active site. Because the PROTAC mechanism depends not just on target:PROTAC interactions, but also on target:E3 ligase interactions that stabilize the ternary complex, it is possible for a PROTAC to mediate degradation of mutated target proteins even when target:PROTAC affinity at the active site is compromised. PROTACs that were designed for wild type targets have been shown to degrade mutant variants 67.
Modularity
PROTACs have a modular construction, comprising two functional ligands bridged by a linker. Each of these 3 components—the target-binding ligand (warhead), the linker, and the E3 ligase ligand—can be systematically varied for efficient optimization of PROTAC physicochemical properties and performance.
PROTAC evolution: from a curiosity to a therapeutic game-changer
First-generation PROTACs, which first emerged in 2001, were large and membrane impermeable, making them impractical for clinical applications. The first membrane-permeable PROTAC was created by adding a cell permeability motif to an E3 ligase binding domain known as the von Hippel Lindau (VHL) peptide. Despite being membrane permeable, these early PROTACs were still of relatively high molecular weight, with poor stability and low potency.
The complex chemistry required for synthesis, as well as issues with membrane permeability, meant that PROTAC technology lay largely dormant until 2015. The first significant breakthrough was the use of very low molecular weight, non-peptidic, VHL mimetics as ligands for the E3 enzyme 8.
Within weeks of this publication, two groups published their work demonstrating that that BRD4 could be successfully degraded with PROTACs based on a thalidomide-based E3 ligand that could target the Cereblon (CRBN) E3 ligase complex 9 10.
These innovations led to the first drug-like PROTACs and proved to be the turning point for transforming PROTAC technology from a novelty research tool into a powerful new drug modality.
Optimization of the first drug-like PROTACs targeting BRD4
Some of the first drug-like PROTACs to be developed were designed to target BRD4, a transcriptional co-activator critical for cancer cell growth and survival.ix,x Since the MYC oncogene had thus far been undruggable using exiting drug modalities, BRD4 was seen as a promising alternative target to inhibit proliferation of MYC-driven cancers such as Burkitt’s Lymphoma (BL) and multiple myeloma (MM). However, the observation that BRD4 inhibitors could inadvertently lead to BRD4 accumulation meant that an alternative approach was needed. With previous evidence that RNAi silencing of BRD4 expression was able to effect a potent antiproliferative response in human and mouse myeloma models 11 12, both groups turned their sights on PROTAC-mediated selective degradation of BRD4, rather than inhibition. Taken together, their results provided compelling evidence that CRBN-based PROTACs could achieve more potent and sustained disruption of target activity, both in vitro and in vivo, compared to conventional small molecule inhibitors.
Again, targeting BRD4 with PROTAC technology, a team at Amgen was the first to publish a comprehensive study of the relationships between PROTAC linker length, E3 ligand choice, ternary complex formation, and protein degradation 13. To accelerate synthesis of candidate BRD4-targeting PROTACs, the group developed “click chemistry” to create a library of PROTACs based on the BRD4 ligand JQ1 in combination with a variety of linkers conjugated to VHL- or Cereblon-based E3 ligase ligands.
The ability of the various PROTAC variants to form a ternary complex was evaluated using AlphaScreen technology, a homogeneous amplified luminescent proximity assay that enabled rapid quantification in 384-well format. The team’s efficient and systematic approach enabled them to progress quickly and find candidates that could successfully degrade BRD4 in two carcinoma lung cell lines. For more detail about their findings and the innovative methodologies used, read PROTAC Assay Design At-A-Glance
PDL1-PROTAC: investigating alternatives to MAb-based therapeutics
Programmed death-1 (PD-1) is an important immune regulator found on the surface of T-cells. When bound to its ligand, PD-L1, this type I transmembrane protein promotes self-tolerance, keeping T cells from attacking other cells in the body, including cancer cells. High tumor expression of PD-1 has been correlated with a high rate of recurrence and unfavorable clinical outcomes (for example see reference 14). Inhibitors that prevent interaction between PD-1 and its ligand are therefore of great interest as anticancer drugs.
In recent years, a number of monoclonal antibodies targeting either PD-1 or PD-L1 have been approved by the FDA. However, such MAb-based drugs are expensive to produce, must be administered intravenously, and have poor bioavailability. Researchers at Bristol Meyers-Squibb therefore investigated PROTACs as an alternative to PD-L1 MAbs 15. Varying both the ligand warhead and the linker (straight or rigid), they synthesized a library of 28 PROTACs, and assessed their ability to function as dual inhibitors and degraders of PD-L1. Homogenous time-resolved FRET (HTRF™), a no-wash proximity-based assay technology, proved to be a rapid and sensitive approach to evaluate PROTAC affinity and the effect of various linker lengths.
The results were instrumental in selecting a PD-L1 PROTAC variant with optimal activity to take forward for further investigation as a potential therapeutic.
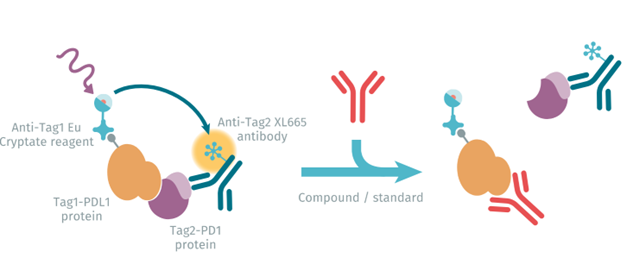
figure 2 : PD1 PD-L1 HTRF assay principle.
A bright future for PROTAC technology
As the first PROTAC-based drugs begin to reach the clinic, the future for PROTAC technology looks bright. PROTAC technology is opening up a world of new possibilities to significantly expand the druggable proteome, develop more potent and selective drugs for cancer and other diseases, and repurpose existing inhibitor candidates that have previously been discounted on the basis of sub-optimal affinity.
Assay design plays an important role to enable accurate and efficient PROTAC characterization and optimization. To learn more about PROTAC applications in oncology and related assay strategies, download our review: PROTAC Assay Design At-A-Glance.
References
- Roskoski Jr R. (2020, September 10). Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Blue Ridge Institute for Medical Research: 62 FDA-approved small molecule protein kinase inhibitors as of 10 September 2020.
- Carles F, Bourg S, Meyer C, and Bonnet P. (2018). PKIDB: A Curated, Annotated and Updated Database of Protein Kinase Inhibitors in Clinical Trials. Molecules 23, 908. DOI:10.3390/molecules23040908.
- Hantschel O. Unexpected off-targets and paradoxical pathway activation by kinase inhibitors. ACS Chem Biol (2015)10: 234-245.
- Raina K and Crews CM. Targeted protein knockdown using small molecule degraders. Current Opinion in Chemical Biology (2017) 39:46-53.
- Sainan A and Fu L. Small-molecule PROTACs: An emerging and promising approach for the development of targeted therapy drugs. EBioMedicine (2018) 36:553-562.
- Buhimschi AD et al. Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry (2018) 57:3564–75.
- Arcila ME et al. EGFR exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther (2013) 12:220–9.
- Zengerle M, Chan K-H and Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 8, 1770–1777.
- Winter GE et al. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science (2015) 348:1376– 1381.
- Lu J et al. Hijacking the E3 ubiquitin ligase Cereblon to efficiently target BRD4. Chem Biol. (2015) 22(6): 755–76.
- J. E. Delmore et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell (2011) 146:904–917.
- J. Lovén et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell (2013) 153:320–334.
- Wurz RP et al 2018. A “Click Chemistry Platform” for the rapid synthesis of bispecific molecules for inducing protein degradation. Journal of Medicinal Chemistry . 61(2): 453-461.
- Jiang F et al. PD-1 high expression predicts lower local disease control in stage IV M0 nasopharyngeal carcinoma. BMC Cancer (2019) 19:503.
- Cheng B. et al. Discovery of novel resorcinol diphenyl ether-based PROTAC-like molecules as dual inhibitors and degraders of PD-L1. European Journal of Medicinal Chemistry (2020) 199:112377.
For research use only. Not for use in diagnostic procedures


