



























Overview
While performing biochemical assays is the most straightforward approach to study protein-protein interactions, oftentimes the cellular environment can be essential to generate the proper interaction. Cell-based assays to study these interactions using Alpha assays can roughly be divided into three categories:
- Alpha assays involving tagged recombinant proteins, transfected into cells: this approach is the preferred one due to the high protein expression levels achieved in transfected cells, and the availability of good anti-tag antibodies.
- Alpha assays involving endogenously expressed proteins: the success of these assays will greatly depend on the endogenous levels of expression of each protein and the availability of specific antibodies directed against the two proteins.
- Alpha assays involving non-tagged recombinant proteins, transfected into cells: while this approach relies on antibody detection, it presents an advantage over the second approach due to higher amounts of proteins expressed in transfected cells.
Studying protein-protein interactions using overexpressed tagged proteins
Selection and orientation of protein tags
Documented cases, showing successful use in conjunction with AlphaLISA™, include FLAG, GST, His and E-Tag (see References below). The use of a protein tag can be especially convenient when working with Alpha technology since the expression of each protein can be confirmed before proceeding to the actual interaction assay. Revvity offers standalone detection reagents (AlphaLISA Acceptor beads and Alpha Donor beads) or complete AlphaScreen® detection kits for these tags (refer to Alpha products and catalog numbers).
Note that the glutathione (GSH) detection system is not recommended for cell-based applications, as lysates contain high concentrations of free glutathione.
The orientation of the selected tags on the recombinant proteins (N- or C-terminal) should be in accordance with current knowledge regarding the interaction of interest such that any possible obstruction toward the interacting regions is minimized. When there is no available information on this, the two proteins should be produced with the chosen tag in both the N- and C-terminal regions for a total of four different products. The different tagging combinations can then be transfected in parallel to find the most optimal one(s), in terms of protein expression.
Generation of expression vector
The protein genes containing the chosen tags should be cloned into appropriate expression vectors.
Transient cell transfection
Many different technologies are available to transfect the expression vectors into cells (Calcium Phosphate, Electroporation, Ballistic Particles, DEAE Dextran, Cationic Matrix, and Lipofection). The lipofection protocol is a relatively simple method that has been used for high throughput screening. FuGENE®6 has been observed to work best in the presence of serum and resulting in little or no toxicity. Below is an example of a transfection protocol for CHO cells using FuGENE®6:
Transfection method for one 100 mm Petri dish (0.5 Million CHO cells seeded 16h before):
- In a 100 mm Petri dish, seed 5 x 106;CHO cells 16 hours before transfection. Place in a CO2 incubator.
- Add 576 µL of serum-free media to a 2 mL sterile polypropylene tube (no antibiotics or fungicide).
- Allow the FuGENE®6 tube to equilibrate to room temperature before opening. Vortex well before opening.
- With a sterile pipet tip, add 9 µL of Fugene6 directly to the serum-free media. Do not touch the sides of the tube. Vortex to mix and wait 5 minutes.
- Add 1-6 µg of DNA to the tube. Wait 15 min.
- Add the transfection mix to the Petri dish containing the cells and growing media in a drop-wise manner over the entire surface. Swirl to mix properly.
- 24-48h post-transfection, harvest the cells and prepare the cell lysate.
We recommend proceeding first with single transfections, in the cell line of your choice, for the two proteins using variable amounts of DNA. This will allow confirmation that each plasmid is producing a functional protein in the cells. Ideally, optimization experiments for DNA amount and post-transfection culture time should be conducted prior to performing double-transfection experiments.
Cell treatment
In certain conditions, treatment of the cells is recommended to achieve an efficient cellular interaction between the two proteins. In some cases, starving the cells may constitute a treatment that will favour the desired protein-protein interaction. In other cases, treating the cells with a known modulator of one of the proteins of interest (agonist, antagonist, inhibitor, etc.) might be an elegant way to generate a control cell lysate. In this example, these would be a ‘’negative’’ (or basal state) lysate and a ‘’positive’’ (stimulated) lysate. It will then be possible to calculate an assay window based on the signals produced with the positive and negative lysates. If no treatments are available, the negative control cell lysate could be a lysate prepared from cells transfected with one protein or the other.
Cell lysis
Lysis buffers
The measurement of protein-protein interactions using Alpha requires the production of cell lysates to release the proteins from their cellular environment. This is a critical step since the lysis per se is a disruptive process involving detergents that could break the protein-protein interaction under investigation. To address that particular need, various non-denaturing formulations of moderate strength have been developed over the years for pull-down and immunoprecipitation assays. The efficiency of lysis buffers to break cells and solubilise proteins will also depend on cell type. We therefore recommend testing several cell lysis buffers. Some that have been successfully used with AlphaLISA are listed below. A protease inhibitor cocktail should be added to all lysis buffers tested. We recommend testing different lysis buffers to select the optimal one.
- 200 mM Tris-HCl pH 7.5, 1% Triton X-100, 50 mM NaCl
- (used for CHO cells, see Waller et al.)
- 100 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.5% NP40.
- (used for BSR T7/5 cells, see Rahman et al. & Mohamed et al.)
- 50 mM Tris-HCl pH 7.5, 125 mM NaCl, 5% glycerol, 0.2% NP40, 1.5 mM MgCl2, 25 mM NaF, 1 mM Na3VO4.
- (used for HEK 293T cells, see Lavens et al.)
- 50 mM Tris-HCl pH 7.5, 0.1% CHAPS.
- (used for CHO cells, unpublished results from Revvity)
We also recommend the following commercially available cell lysis buffers:
- AlphaLISA lysis buffer (Revvity, Cat. No. AL003)
- (proprietary formulation)
- Cell lysis buffer (10x) (Cell Signaling Technology, Cat. No. 9803)
- (1X Cell lysis buffer: 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/mL leupeptin)
- M-PER® Mammalian protein extraction reagent (Thermo Scientific, Cat. No. 78501)
- (proprietary formulation)
- Pierce IP lysis buffer (Thermo Scientific, Cat. No. 87787)
- (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol)
Preparation of lysis buffer
- Dilute the lysis buffer to 1X (if needed) prior to use.
- Add a standard protease inhibitor cocktail (Sigma Cat. No. P2714 or Roche Cat. No. 05 892 791 001) to the lysis buffers prior to use. Reconstitute according to the manufacturer’s instructions. Other cocktails can be used as well.
Cell lysis procedure
- Treat the cells if needed.
- Detach the cells if required.
- Determine the cell density of the culture. Harvest the cells by centrifugation.
- Discard the supernatant and resuspend the cells in 1X Lysis buffer. The optimal number of cells must be determined experimentally. However, 4 x 106 cells/mL are recommended as a starting point. Add 5 µL of 1X Lysis buffer to 20,000 cells.
- Incubate for about 5 - 10 minutes at room temperature on a rocker to maximize the lysis.
- Centrifuge for 5 minutes at high speed (13,000 RPM) in a benchtop centrifuge to clarify the lysate. Discard the pellet.
- Prepare aliquots and freeze at -80 °C.
Protein expression assessment
Once the bulk cell lysates are prepared, as a preliminary step it is important to evaluate the relative levels of expression of each protein in the cells. We cannot stress enough the importance of gathering solid evidence for expression of the respective proteins before proceeding to the actual detection of the interaction. Although this does not guarantee success in measuring a protein-protein interaction, it is certainly the most important step towards that goal. Examples and recommendations using Alpha assays are given below, but other methods may also be used.
Tag-based detection with Alpha technology
Typically, the confirmation of protein expression in a cell lysate can be done by performing a competition-type assay set up to detect a tagged positive control peptide included in an AlphaScreen kit (for example, a biotinylated and flagged peptide). To this assay are added increasing amounts of lysate containing the expressed protein bearing the expression tag (see Figure 1 below). A signal decrease proportional to the amount of cell lysate is expected, as the expressed protein can compete with the positive control. This step should be done for each protein. If necessary, the amount of DNA transfected can be adjusted to achieve the desired expression levels. Non-transfected cells or cells transfected with an unrelated target should be included as negative controls to rule out any interference coming from non-specific components of the lysates.
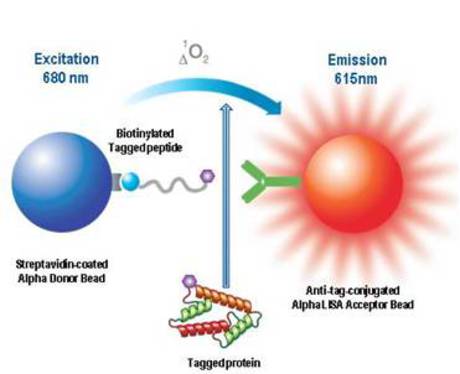
Figure 1. Schematic diagram of an assay used to confirm cellular expression of the tagged protein. The overexpressed tagged protein competes for binding of biotinylated tagged peptide to anti-FLAG conjugated Acceptor beads.
Figure 2 is an example where the expression of a FLAG-tagged protein was confirmed in transfected CHO cells. The assay was performed in a 384-well OptiPlate™ for a 25 µL final assay volume according to the following procedure.
- Add 10 µL Anti-FLAG Acceptor Beads (20 µg/mL final) diluted in assay buffer.
- Add 5 µL CHO lysate dilutions in lysis buffer.
- Incubate 15 min at room temperature.
- Add 5 µL of biotin-FLAG peptide (5 nM final) in assay buffer. Incubate 15 min at room temperature.
- Add 5 µL of streptavidin Donor beads (20 µg/mL final) diluted in assay buffer.
- Incubate 30 min at room temperature.
-
Read on an Alpha-capable reader (EnVision® Multilabel Plate Reader or EnSight™ Multilabel Plate Reader).
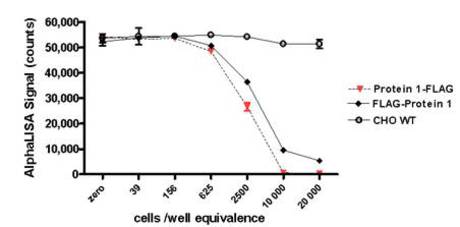
Figure 2. Detection of a FLAG-tagged protein in two transfected lysates using an Alpha assay. The detection of FLAG-tagged Protein 1 (with either an N- or C-terminal tag) was done using an AlphaScreen FLAG detection kit. Serial dilutions of the lysates were tested for their ability to compete with the biotin-FLAG peptide. The amounts of lysate are reported in cells/well equivalence (20,000 corresponding to the undiluted lysates). Untransfected CHO WT lysates were included as a negative control. Here, the Protein 1-FLAG transfect (C-terminal FLAG tag) displayed a slightly higher expression level compared to the FLAG-Protein 1 transfect (N-terminal FLAG tag), indicating that the FLAG tag in the C-terminus may be more optimal.
In a similar fashion, the expression of Protein 2 tagged with GST could be confirmed using Anti-GST Acceptor beads and the same Anti-GST antibody biotinylated and captured by streptavidin-Donor beads (Figure 3). Note that the Anti-GST antibody is polyclonal, which allows a sandwich capture of the GST protein. As a result, the signal increases with increasing target protein concentrations. The assay was performed in a 384-well OptiPlate for a 25 µL final assay volume.
- Add 10 µL Anti-GST Acceptor Beads (20 µg/mL final) diluted in assay buffer.
- Add 5 µL CHO lysates dilutions in lysis buffer.
- Incubate 15 min at room temperature.
- Add 5 µL of biotin-Anti-GST (1 nM final) in assay buffer.
- Incubate 30 min at room temperature.
- Add 5 µL of streptavidin Donor beads (20 µg/mL final) diluted in assay buffer.
- Incubate 15 min at room temperature.
-
Read on an Alpha-compatible reader (EnVision or EnSpire Multilabel Plate Readers).
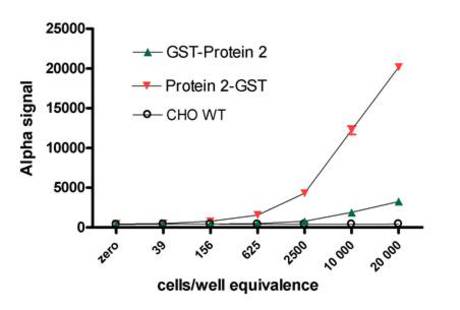
Figure 3. Detection of a GST-tagged protein in two transfected lysates using an Alpha assay. The detection of GST-tagged Protein 2 (with either an N- or C-terminal tag) was done in a sandwich assay involving Anti-GST Acceptor beads and a biotinylated goat Anti-GST antibody. Serial dilutions of lysates were tested in the sandwich assay. The amounts of lysate are reported in cells/well equivalence (20,000 corresponding to the undiluted lysates). Untransfected CHO WT lysates were included as a negative control. Results indicated that expression levels were distinctly higher when the GST tag was located at the C-terminal end of Protein 2.
Additional ways to assess protein expression
The experimental examples given here can help in confirming the expression of the tagged proteins. However, this methodology does not constitute a definitive proof that the cell expresses a fully functional tagged protein. For example, the protein may be truncated or cleaved and the tag could not be attached to the protein. We therefore suggest performing a Western blot detection to verify that the expressed tagged proteins display the expected molecular weights.
If an antibody is available to the tagged protein, one could use an Alpha assay where the tagged protein is detected using both the tag and a specific antibody in a capture assay format. Compared to the tag-based detection strategy described above, this type of assay can provide more compelling proof for the functional expression of the tagged proteins since the antibody epitope and the tag need to be present on the same protein to generate an Alpha signal.
Based on the results obtained in this section, the best constructs and the optimal lysis buffer should be selected for use in subsequent double-transfection and protein-protein interaction experiments.
Alpha protein-protein interaction assay
When you are ready to perform the double-transfection experiments for the actual detection of an interaction, we suggest that various ratios of DNA be tested in transfections since a 1:1 stoichiometry in DNA amount will not necessarily result in comparable expression levels for the two proteins. Various factors may affect the efficiency of transfection, protein transcription and maturation.
Selection of assay buffer
While there is no universal assay buffer that can be recommended to suit all protein-protein interaction studies, the following two buffers were shown to be successful in some cases:
- PBS + 0.1% BSA (ref. Rahman et al.; Mohamed et al.)
- 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1% BSA (Revvity unpublished results)
In addition, test any assay buffers that have been documented in the literature for use with the protein-protein interaction of interest. One general recommendation is to avoid the use of detergents in the assay buffer composition. Some detergent will most likely be introduced to the assay by the sample. (The sample lysis buffer will be diluted 5-fold if 5 µL of sample is added in a 25 µL assay.) Excessively high levels of certain detergents could affect the protein-protein interaction and the Alpha technology as well. Also it is important to verify that the buffer does not contain substances known to interfere with the Alpha technology. The tolerance of Alpha technology to various detergents and other substances is detailed in Buffer selection for Alpha assays.
Confirmation that a positive signal is a valid protein-protein interaction
Once the double transfections have been performed at various DNA ratios and cell lysates have been prepared, the validity of the protein-protein interaction assay should be confirmed. One or more of the following approaches may prove useful in validating that a positive Alpha assay signal from double-transfected cell lysates is indeed due to the desired protein-protein interaction.
- As negative controls, test single-transfected lysates in the protein-protein interaction assay.
- To confirm that the signal observed correlates with the amount of double-transfected cell lysate, perform a titration of the amount of double-transfected lysates added to the assay (see Figure 4, below).
- To confirm that the signal observed is due to the presence of the two specific proteins, when possible, perform a competition experiment in which increasing concentrations of a recombinant form of one of the two proteins is added to the cell lysates (see Figure 5, below).
- To further confirm the specificity of the Alpha assay signal, reference compounds or peptides known to interfere or modulate the protein-protein interaction can be tested for their effect on the assay.
Titration of double-transfected Cell lysates in Alpha assays
A cell lysate titration experiment should be performed on each of the double-transfected cell lysates. The assay should be set up detect the interaction of the two overexpressed proteins under study. Serial dilution of the lysate should correlate with a decreasing signal in the assay. Ideally, a negative control should also be performed using lysates of non-transfected cells or cells transfected with a non-related target cDNA.
Figure 4 shows the results of a serial dilution of lysate from a cell transfected with one ratio of Protein 1-FLAG / Protein 2-GST DNA. The Alpha assay employed a biotinylated anti-GST antibody and anti-FLAG-conjugated Acceptor beads. In this example, the data confirmed that the amount of signal correlated with the amount of lysate added. The assay was performed in a 384-well OptiPlate with a 25 µL final assay volume using the following protocol.
- Add 10 µL of CHO lysates prepared in lysis buffer.
- Add 5 µL Anti-FLAG Acceptor beads (20 µg/mL final) diluted in assay buffer.
- Incubate 30 min at room temperature.
- Add 5 µL Biotinylated Anti-GST antibody (1 nM final) diluted in assay buffer.
- Incubate 60 min at room temperature.
- Add 5 µL streptavidin Donor beads (20 µg/mL final) diluted in assay buffer.
- Incubate 60 min at room temperature.
-
Read in Alpha-compatible reader (EnVision or EnSpire Multilabel Plate Readers)
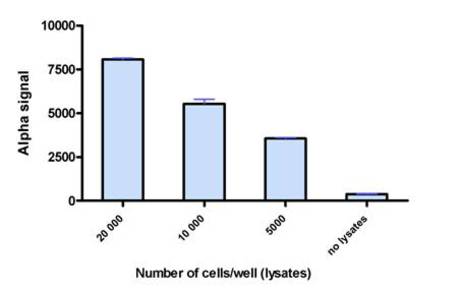
Figure 4. Alpha assay detection of tagged Protein 1-Protein 2 interaction, illustrating that the signal correlates with varying amounts of protein-protein complex. Lysate dilutions prepared from cells transfected with Protein 1-FLAG / Protein 2-GST DNA were tested in an Alpha assay. AlphaScreen Anti-FLAG Acceptor beads and streptavidin Donor beads were used at 20 µg/mL final, and biotinylated anti-GST was used at 0.3 nM final.
Competition protocol to validate protein-protein interaction assay
Where a recombinant form of one of the target proteins is available, this can be added to the double-transfected cell lysate in increasing concentrations (Figure 5). A reduction in signal with increasing concentrations of this competing protein suggests that the assay is detecting the desired protein-protein interaction. One essential requirement for such a competition experiment is that the recombinant protein must not contain either of the two tags used in the detection assay (FLAG and GST in this example). The assay shown here was performed as follows in a 384-well OptiPlate using a 25 µL final assay volume.
- Add 5 µL Anti-FLAG Acceptor Beads (20 µg/mL final) diluted in assay buffer.
- Add 5 µL recombinant Protein 2, diluted in assay buffer.
- Add 5 µL CHO lysates (prepared at 4M cells/mL).
- Incubate 60 min at room temperature.
- Add 5 µL of biotin-Anti-GST (1 nM final) diluted in assay buffer.
- Incubate 60 min at room temperature.
- Add 5 µL of streptavidin Donor Beads (20 µg/mL final) diluted in assay buffer.
- Incubate 60 min at room temperature.
-
Read on an Alpha-compatible reader (Envision or EnSpire Multilabel Plate Readers).
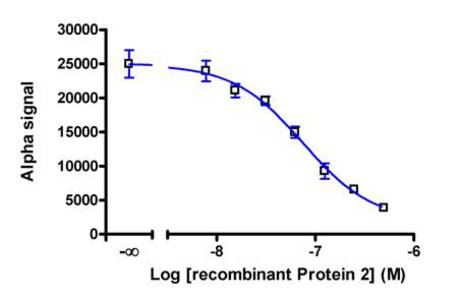
Figure 5. Competition assay performed to validate a protein-protein interaction assay for Protein 1-FLAG and Protein 2-GST. Lysates prepared from CHO cells double-transfected with Protein 1-FLAG and Protein 2-GST DNA were tested in the presence of increasing concentrations of recombinant Protein 2. An IC50 value of 73 nM was calculated.
Studying protein-protein interactions using endogenous proteins
The detection of an interaction between endogenously expressed proteins must rely on the use of specific antibodies, since the proteins will not have expression tags. As for the tagged overexpressed proteins discussed in the previous section, it is crucial to gather good evidence that the two endogenous proteins to be studied for interaction are indeed expressed in the cellular model chosen.
In the case of endogenous non-tagged proteins, the most popular technique for achieving this is the Western blot. One can also use an antibody-based Alpha assay to detect each protein individually. In this instance, a variety of antibodies should be tested in order to maximize the chance of finding at least one robust pair of antibodies for the assay.
When data from ELISA experiments are available, one should first select the same pair of antibodies to try in an Alpha assay. If this is not successful, other antibody pairs can then be investigated.
Antibody selection
The selection of antibodies should be made based on the available literature for the interaction. The chosen antibodies should bind to an epitope that is as distant as possible from the interacting domain(s) of the proteins to avoid steric hindrance. When the map of the interaction is unknown, we suggest selecting and testing multiple antibodies that target different regions of the protein. Combinations of these antibodies can then be tested in a matrix experiment to find the optimal pair. A practical search engine for commercially available antibodies can be found in the Biocompare® site.
When available, recombinant versions of the studied proteins could prove helpful in selecting antibodies and possibly in confirming the specificity of the putative protein-protein interaction. When testing different antibody pairs in an Alpha assay, the assay can be relatively quickly developed using a recombinant protein as the target. Once the protein-protein assay format has been established, the recombinant protein can be used to show assay specificity by competing with the interaction of the endogenous proteins being studied (see section above) for an example of this approach using overexpressed proteins)
Antibody labeling
All of the selected antibodies should be prepared for use in both of the possible orientations in the Alpha assay. In other words, an aliquot of each antibody should be biotinylated (for capture on streptavidin Donor beads) and a separate aliquot should be directly coupled to Acceptor beads.
Cell lysis
Please consult the Cell lysis section above for detailed information on cell lysis.
Protein expression assessment
Once the bulk cell lysates are prepared, a preliminary step consists in evaluating the relative level of expression of each protein in the cells. This can be done in an Alpha assay by capturing each individual protein with specific antibodies or by Western blot analysis. Different lysis buffers may be tested at this stage. As for tagged proteins, it is important to verify that the assay signal is specific for the targeted protein and is correlated with the amount of lysate added to the assay.
Alpha interaction assay for endogenous proteins
Matrix experiments
The initial search for a positive signal in an Alpha assay demonstrating a protein-protein interaction event will involve exploring two important parameters: lysis buffer and antibody selection. The first experiments should consist in matrix assays where all the combinations of the various lysis buffers and antibody pairs will be tested in parallel (Figure 6).
For each antibody pair tested, we recommend including a negative control for which no lysate is added (lysis buffer only). Alternatively, if the interaction is known to be modulated by a cell treatment, treated versus untreated cell lysates might serve as positive and negative controls. This should help discard any potential cross-reactivity or non-specific signal that could mistakenly be thought to originate from the interaction occurring between proteins.
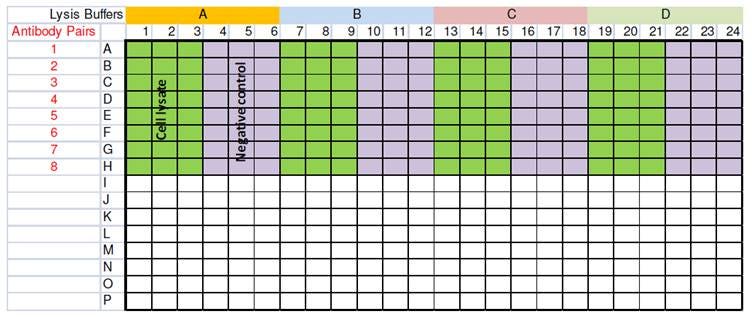
Figure 6. Example of a matrix assay for selection of a lysis buffer and antibody pairs. In this 384-well microplate setup, four lysis buffers and eight antibody pairs are tested in triplicate.
Confirmation that a positive signal is a valid protein-protein interaction
Refer to section above for examples of experiments that can confirm the validity of your protein-protein interaction Alpha assay.
Studying protein-protein interactions on non-tagged recombinant proteins
The approach taken here is a combination of the fundamentals covered in the above sections. It relies on specific antibodies directed against the recombinant proteins, and, for that reason, these assays are more challenging than those involving tagged recombinant proteins. On the positive side, they are in general easier to develop than the assays for endogenous proteins due to the higher expression levels of recombinant proteins.
See sections above for information on preparing the cell lysates for analysis. As for other applications, lysates should be prepared and tested from cells expressing each individual protein before working with lysates from cells coexpressing the two binding partners.
When the proteins to be studied do not include any tag, the confirmation of expression and selection of the best lysis buffer can be made using techniques such as Western blotting. It is also possible to create Alpha assays for detecting each protein individually by using two specific antibodies in a sandwich assay format. Performing these validation assays in an Alpha assay format may require some additional time and effort initially, but will ultimately save hands-on time as compared to Western blotting. These benefits can prove especially useful when handling a large number of experimental conditions (e.g. optimization of transfected DNA amounts for each protein).
See section above for help with setting up the Alpha protein-protein interaction assay.
References
Literature examples of Alpha cell-based assays using recombinant and tagged proteins:
| Reference | Tags used | Lysis buffer | Lysis protocol | Assay buffer | Lysate amount |
|---|---|---|---|---|---|
| Becker et al. | FLAG and HA | 100 mM Tris, pH 8.0, 100 mM NaCl, 0.5% NP-40, 0.2 mM PMSF containing Roche complete protease inhibitor | Twenty-four hours post-transfection, cells from one well of a 6-well microplate were rinsed with PBS and lysed on ice for 5 minutes. The cellular extracts were then separated following centrifugation for 10 min at 13,000 g at 4°C. | PBS + 0.1% BSA | 2 µL of lysate from one well was diluted in 1:10 in assay buffer. |
| Rahman et al. | GST and His | 100 mM Tris pH 8.0, 100 mM NaCl, 0.5% NP-40, containing Roche complete protease inhibitor | Forty-eight hours post-transfection, cells were collected in 500 mL PBS, pelleted and then lysed by suspending in 20 mL lysis buffer. The cellular extracts were then separated following centrifugation for 5 min at 13,000 rpm. | PBS + 0.1% BSA | Cells seeded in 24 well plates. Forty eight hours post transfection, cells were collected in 500 mL PBS, pelleted and then lysed by suspending in 20 μl lysis buffer. 5 μl of this cell extract were mixed to a total assay volume of 25 μl. |
| Lavens et al. | Etag and FLAG | 50 mM Tris-HCl pH 7.5, 125 mM NaCl, 5% glycerol, 0.2% NP40, 1.5 mM MgCl2, 25 mM NaF, 1 mM Na3VO4, Complete protease inhibitor without EDTA cocktail. Lysates were cleared by centrifugation. | not disclosed | not disclosed | |
| Mohamed et al. | His and GST | 100 mM Tris pH 8.0, 100 mM NaCl, 0.5% NP-40, containing Roche complete protease inhibitor | Forty eight hours post transfection, cells were collected in 500 mL PBS, pelleted and then lysed by suspending in 20 mL lysis buffer. The cellular extracts were then separated following centrifugation for 5 min at 13,000 rpm. | PBS + 0.1% BSA | Cells seeded in 24 well plates. Forty eight hours post transfection, cells were collected in 500 mL PBS, pelleted and then lysed by suspending in 20 μL lysis buffer. 5 μL of this cell extract were mixed to a total assay volume of 25 μL. |
| Werden et al. | His, FLAG and HA | Not disclosed | Not disclosed | PBS + 0.1% BSA | Not mentioned |
| Waller et al. | FLAG, GST, His and HA (His and HA for transfected cells) | Following a PBS wash: 200 mM Tris-HCl pH 7.5, 1% Triton X-100, 50 mM NaCl | The cell lysates were incubated for 30 min on ice and then centrifuged at 9,357×g in a microcentrifuge for 5 min at 4°C. | 100 mM HEPES pH-7.5, 1mM EDTA, 5mM DTT, 0.1% CHAPS, 5% glycerol. | CHO cells grown in 6 well plate, transfected and grown for 48 h then lysed in 100 μL. 18 μL of this lysates is used per well of Alpha. |
| Internal data presented on this page | FLAG and GST | Bulk produced at 4M/ml in 50 mM Tris-HCl pH 7.4, 0.1% CHAPS + protease inhibitor cocktail (note: CHAPS was documented as a detergent that preserves the studied interaction). | 50 mM Tris-HCl pH 7.5, 0.1% CHAPS | 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% BSA | 5 μL of 4M/mL lysates in 25 μL total assay volume |
For research use only. Not for use in diagnostic procedures. The information provided above is solely for informational and research purposes only. The information does not constitute medical advice and must not be used or interpreted as such. Consult a qualified veterinarian or researcher for specific guidance or use information. Revvity assumes no liability or responsibility for any injuries, losses, or damages resulting from the use or misuse of the provided information, and Revvity assumes no liability for any outcomes resulting from the use or misuse of any recommendations. The information is provided on an "as is" basis without warranties of any kind. Users are responsible for determining the suitability of any recommendations for the user’s particular research. Any recommendations provided by Revvity should not be considered a substitute for a user’s own professional judgment.